蛋白质组学是一项涉及蛋白质表达谱分析、蛋白质结构测定、蛋白质翻译后修饰(PTMs)、蛋白质功能分析和蛋白质-蛋白质相互作用,在人类基因组中被发现的蛋白质编码基因只有2万个,而蛋白质编码基因的总数超过了100万个。
液相色谱-质谱法(LC-MS)是研究高通量蛋白质组学的首选方法,传统的单维LC的分离性能受到峰容量的限制,阻碍了蛋白质组学的发展。
可以将不同的分离模式正交组合,开发出高峰容量的多维液相色谱(MDLC)。
基于mdlc-ms的血浆蛋白质组学新策略完整蛋白质的氨基酸序列和翻译后修饰等重要信息在消化过程后可能缺失,特别是低丰度的目标蛋白质。
同时,随着蛋白水解肽种类和数量的增加,样品的复杂性也随之增加,更难以追溯相同肽段的蛋白质。
通过开发了一种基于阵列的在线2D-Strong阴离子交换(SAX)-RPLC系统,用于分离人血浆蛋白和消耗高丰度蛋白。第一维分离采用强阴离子交换柱,第二维分离采用8个阵列RP柱。
在此条件下,与传统的二维液相色谱法相比,该阵列法的效率和通量均提高了8倍。
通过对实际血浆样品的分析,共鉴定出84个高丰度蛋白质,1332个蛋白质,回收率超过95%。检测到的蛋白浓度范围为0.01mg/mL~41mg/mL,涵盖9个数量级。
因此,基于阵列的多维液相色谱在分离效率方面具有很大的优势,但仪器较为复杂,在大规模蛋白质组学分析中具有广阔的应用前景。(图1)

(图1)
还有另一种方法是基于spintip的简单集成的蛋白质组学技术(SISPROT),由于操作相对容易、学习曲线短,已成为基于mdlc-ms的蛋白质组学方法的重要解决方案之一。
并且基于spin-tip的MDLC-MS方法,用于快速和深入的血浆蛋白质组分析,是混合模式-sisprot,使样品操作和分馏步骤,包括蛋白质消化,SCX/SAX,高ph反相分馏首次集成到一个单一的自旋尖端设备。
在保持最小样品损失和分离效率的情况下,在2小时内实现深度血浆蛋白质组学分析,并可在生理pH条件下加载血浆样品。从1μL高丰度蛋白质耗尽(约60μg)的血浆中鉴定出多达862种蛋白质。
这种新型的血浆蛋白质组分析方法可以在生理条件下进行样品制备,具有重要的生物和临床应用潜力,特别是对珍贵或少量的样品。
更深层次的序列覆盖2D-SAX-RPLC方法可以通过更有效的分离,对人尿蛋白进行更深层次的序列覆盖。
优化后,2D-LC系统的理论峰值容量超过40000,与单一分数一维方法鉴定出的628个蛋白质相比,最多鉴定出2440个蛋白质。(图2)

(图2)
同时,两种方法共鉴定了588个蛋白质,2D分离方法显著提高了蛋白质序列的覆盖度,讨论了不同氨基酸数蛋白质的序列覆盖度。对于小于500的AA编号,序列覆盖率提高了2.02倍。
与传统的1D方法相比,随着AA数的增加,序列覆盖水平有所提高。当AA数超过1000时,序列覆盖度增加了约3.87倍。
为了破译蛋白质的c端序列,开发了一种基于2D-SCX-RPLC-MS/ms的方法,即BaSCX碱性强阳离子交换色谱(BaSCX)。
结合LysargiNase蛋白消化和高pH条件下SCX分离能力,通过两小时的LC-MS/MS分析,从40μghela细胞裂解液中鉴定出2151个数据库注释蛋白c-端和166个neoc-端。
相比之下,数量传统胰酶-消化法鉴定出的c-末端从未超过1000。
该方法解决了长期存在的蛋白质c-末端分析技术难题,更多的研究报道了基于MDLC的深度序列覆盖,表明MDLC结合质谱具有更好的分离效率,大大降低了样本复杂度,提高了蛋白质组学研究中蛋白质序列的覆盖深度。
这对于建立更精确的蛋白质组数据库和更好地理解生命过程至关重要。
蛋白质复合物分析传统的基于抗体的方法仍然存在非特异性免疫亲和力问题,将背景蛋白误认为相互作用蛋白,可能导致获得的结果可信度低,具有误导性,基因编辑策略的方法往往是复杂的手动操作,这是相当耗时和需要沉重的工作量。
蛋白质相互作用(protein-proteininteraction,PPI)分析可能受到细胞类型和空间位置的限制,该方法具有分离能力强、通量大、自动化程度高、灵敏度高的特点,在蛋白质复合物的分离和分析方面具有广阔的应用前景。
通过设计并建立了基于阵列的在线2D-LC-MS平台,首次将MDLC-MS策略应用于蛋白质-蛋白质相互作用的分析。
第一维为正交大孔混合床离子交换柱,第二维为8个自制RPLC分离柱阵列,结合Q-Exactive质谱仪,对蛋白质复合物进行彻底分离后的分析。
全自动化和基于阵列的分析策略可以大大减少蛋白质复合物的分析时间,分离效率比传统在线MDLC方法提高了8倍。
由于没有实施人工操作过程,实现了高重现性,消除了任何人为样品污染的风险。
所以从Hela裂解液中预测出了47,745个蛋白质-蛋白质相互作用和1530个具有高可信度的假定蛋白质复合物,是目前从海拉细胞获得的最大的推定蛋白质复合物数据库,提高基于阵列的MDLC-MS/MS分析系统为大规模的高通量蛋白质相互作用体研究效率。
基于sec的LC-MS方法具有样本大小分离效率高、分辨率有限的特点,具有分离蛋白质复合物等较大目标的优势。采用一种新颖的2D-SEC-RPLC方法,用于大规模蛋白质复合物分析。
将SEC与基于针尖的高pH反相(HpHt)分离相结合,可以提高交联肽的分离效率和纯度。结合交联质谱技术(XL-MS),对蛋白质复合物有了更深入的了解。
从人类HEK296细胞裂解液中鉴定了10,932个独特的交联肽,从数据集中鉴定了780个CORUM蛋白复合物,分布在涉及核糖体、rna前剪接体等多个亚细胞结构中。所以说基于mdlc的高通量、高分离能力的方法将在大规模PPI分析中发挥更大的作用。(图3)

(图3)
MDLC自动化的进展-小型化通过mdlc-ms基础方法的自动化和小型化,可以实现对微克甚至纳克样品的全面原型分析。
比如一种全自动化的纳米流2D-RP-RPLC-MS系统nanoFAC。在75μ、流速为300nL/min的毛细管柱中,以高pHRPLC作为第一个维度,采用自动分数级串联。用自动进样器将第1维的馏分转移到第2维以进一步分离。(图4)
减少样品损失的措施和分级收集容器材料会样品回收的影响,0.01%n-十二烷基β-d-麦芽糖苷(DDM)在部分缓冲液中显著加深了蛋白质组的覆盖范围,识别出的磷酸化多肽数量增加了约45%。
而与玻璃小瓶相比,聚丙烯材料作为样品收集的容器显著减少了样品损失,肽和蛋白质回收率分别提高了370%和110%。
这样有助于进一步减少样品在分离和转移过程中的损失,从而减少总样品量,实现了从100ng蛋白质样品中6000多个蛋白质的综合鉴定。
当与磷酸化肽富集方法相结合时,从100μgMCF-7细胞裂解液中鉴定出2万个磷酸化肽,比常规一维nanoLC增加了144%,所以2D-LC-MS系统的小型化和全自动化以及进样-制备工艺的优化等措施可以显著减少样品损失,为未来的单细胞MDLC分析提供了可能性。
还可以采用一种低流量在线2D-HILIC-RPLC-MS方法,第一维选用国产ZICHILIC毛细管柱,第二维选用C18反相柱。采用稀释主动调制技术解决了溶剂的配伍问题。
在不增加分析时间的情况下,小型化的2DLC系统减少了所需的样品总量,同时增加了60%的峰容量和17-34%的蛋白质数量。
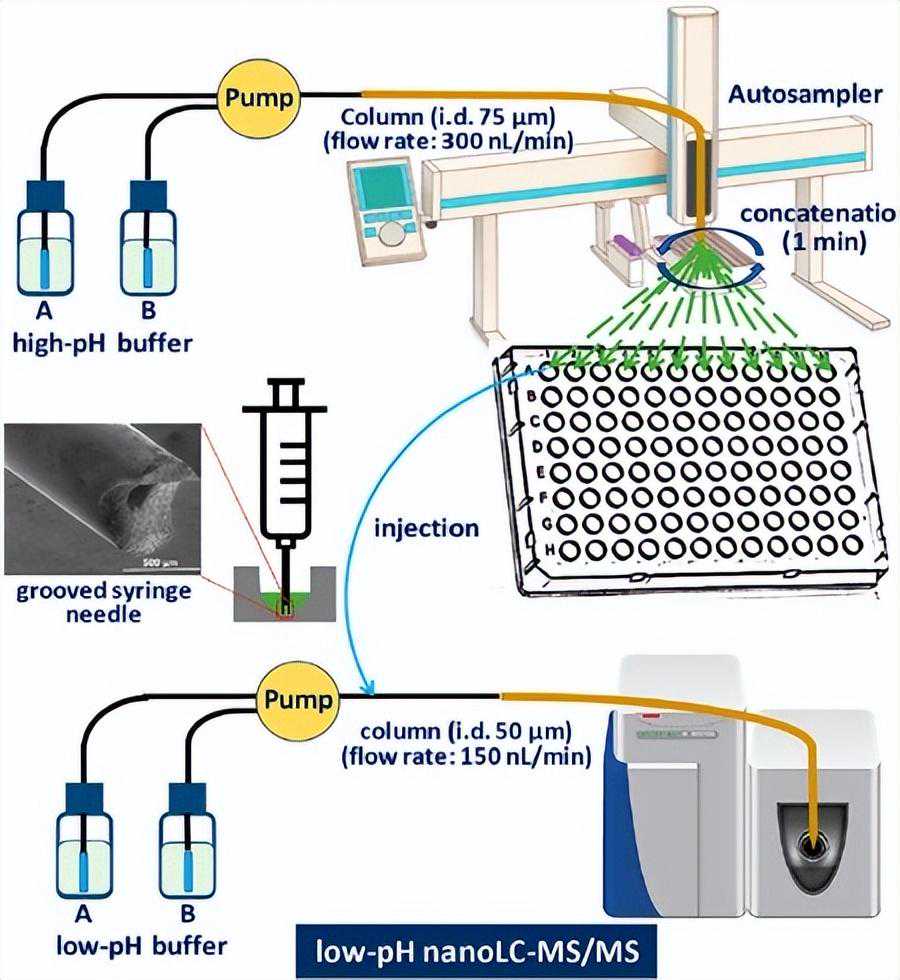
(图4)
无人化样品处理方法处理过程的自动化将最大限度地减少人工对样品的影响,通过在线、自动化的2DNanoMDLC-MS粗血清蛋白质组分析系统避免了可能导致系统误差的预处理过程和人为操作,使三个独立的实验室通过相同的实验过程获得一致的结果。
虽然总处理量降低了4倍,但总多肽数量增加了60%,总样品量明显减少到3μg,主要是由于简化的样品预处理和自动化的工作流程,更深入的蛋白质组覆盖和可接受的重现获得了延展性,这对多中心、大队列研究至关重要。
采用演示了一种名为“μSEC2-nRPLC-MS”的2DLC-MS溶液用于完整蛋白分析,自行设计的进样量控制(IVC)策略,在SEC柱前后插入蛋白陷阱柱,实现了大容量进样。
2DLC-MS采用高通和低通SEC过滤作为第一维,脱盐和净化过程均在全自动化条件下完成。
分子量在30kDa以下的完整蛋白被富集和纯化,同时消耗高分子量的血清蛋白,总样品量减少了100倍。与一维LC相比,大约10倍以上的变形体被识别。
这种自动化的在线2D-LCMS方法为完整的低分子量蛋白质的大规模蛋白质组学研究提供了一个可选的技术路线,并具有筛选低分子量蛋白质生物标志物的潜力。(图5)

(图5)
其他基于mdlc-ms的蛋白质组学研究方法在线2D-SCX-RPLC系统MudPIT通过将强阳离子交换和反相材料填充到一个双相微毛细管柱中,与下游的质谱具有良好的兼容性。MudPIT的出现极大地推动了大规模蛋白质组学的发展。
然而传统的MudPIT方法仍然存在SCX相的淋溶盐问题,因为淋溶盐不仅会抑制检测信号,还会降低MS的性能,甚至损坏MS仪器。
现有一种改良版的MudPIT,即所谓的平行通道-多维蛋白质鉴定技术(PC-MudPIT)。采用两个平行分析柱作为该方法的主要改进和亮点。
通过切换柱的位置,scx-洗脱过程下的柱在流道中远离质谱仪的入口。分析柱经纯水脱盐后,转至质谱入口,准备进行二次反相分离。
长期存在的盐污染和洗脱导致的分析效率相对较低的问题,而传统泥浆坑的所有优点都得到了继承。然而,管道长和死容积大等缺点仍然需要解决。
还有另一种改进的12步MudPIT和一种新开发的定量BONCA(QBONCAT)质谱技术来分析和定量视神经损伤后视网膜中新合成的蛋白质。
在叠氮同丙氨酸(Azidohomoalanine,AHA)click化学反应中,利用生物素的同位素变异,BONCA定量检测了1792个新合成的蛋白质及其在视神经损伤后第1天或第5天的转运率变化。(图6)
此外,在已有的MDLC方法中,采用了多种固定相填料,包括一些新报道的填料或混合填料。
例如在一维上应用了商业化的三模态相填料(由反相、弱阴离子交换和强阳离子交换材料组成),在温和酸性条件下,利用乙腈梯度实现了共有肽和tmt标记肽的分离。
可接受的正交性和与质谱系统的直接兼容性得到保证,大大减少了样品和某些特殊的PTMs的损失。
虽然这些尝试可能导致更好的分离结果,但即使在相同的分离条件下,分析相同的目标时,保留行为和相应的保留时间也可能发生改变。
因此,建立可靠的保留时间预测模型对于更好地理解多肽在不同固定相下的分离模式和机制,从而促进创新MDLC技术的发展至关重要。
目前已经建立了HILIC、SCX、SAX等分离模式的肽滞留时间预测模型,系统地定量研究了可能的分离机制、影响因素以及序列特征对滞留行为的影响。

(图6)
人们还发现了新的分段二维液相色谱(S2DLC)方法,1维空间分离与2维时间分离的综合在线结合。
开发了一个理论模型来描述和预测分析物在第一维的位置和在第二维的洗脱时间,并成功地将该方法及其模型应用于分析尿源性神经递质。
此外,基于3dlc-ms的方法已显示出其在综合蛋白质样品分析方面的潜力,但“基于时间序列”的顺序分离方法受到长时间分析时间的限制。
“天基”MDLC可能提供一种替代解决方案,它结合了较短的分析时间和相对较高的峰值容量。
通过设计了一种空间3DLC系统,可以保证从第二维度和第三维度同时分离分数。
样品先在一维通道中分离,然后通过流量分配器垂直转移到第二维通道,再转移到第三维通道。虽然在保证固定相在不同分离区域的均匀、正交叠加、流速控制、样品检测等方面还存在一些技术问题,但解决上述问题的尝试已经证明它具有空间3DLC实现的潜力。(图7)

(图7)
基于mdlc-ms的方法优势在于其高峰容量所赋予的高分离能力,在蛋白质组学研究中发挥了重要作用。
通过正交选择最佳的分离模式,并结合高灵敏度和多路复用质谱技术,MDLC为不同情况下的综合和靶向蛋白质分析提供了一个强大的工具箱。
此外,基于MDLC-MS的全自动方法为大规模、高通量的蛋白质组学研究提供了可选的解决方案,有利于MDLC-MS方法的推广,将进一步促进蛋白质组学特别是临床蛋白质组学研究的发展,未来的研究将继续集中在高度通信的发展上兼容高效的在线MDLC-MS技术。